软件使用
4.1 基本使用
1. 准备输入数据的格式
SoCube的数据输入是scRNA-seq的UMI计数矩阵,其行为细胞,列为基因,值为UMI计数。SoCube接受两种数据格式:(1)Python pandas库保存的HDF5格式DataFrame对象;(2)Python anndata库保存的H5AD格式(一种定制化的HDF5格式)AnnData对象。从事单细胞组学领域研究的人都具有一定的数据处理基础,因此本手册不具体介绍如何从其他格式转化为上述格式。
2. 运行SoCube
SoCube和前面提到的pip一样,运行于终端下,这样的优势在于可以运行在很多没有GUI页面的服务器上,而不受GUI限制。假设输入文件为"D:\data\pbmc-1C-dm.h5ad",则可以在终端输入下面命令运行软件,然后就会看到软件启动并在终端输出日志。参数-i(或者--input)表示指定输入文件。
socube --input "D:\data\pbmc-1C-dm.h5ad"
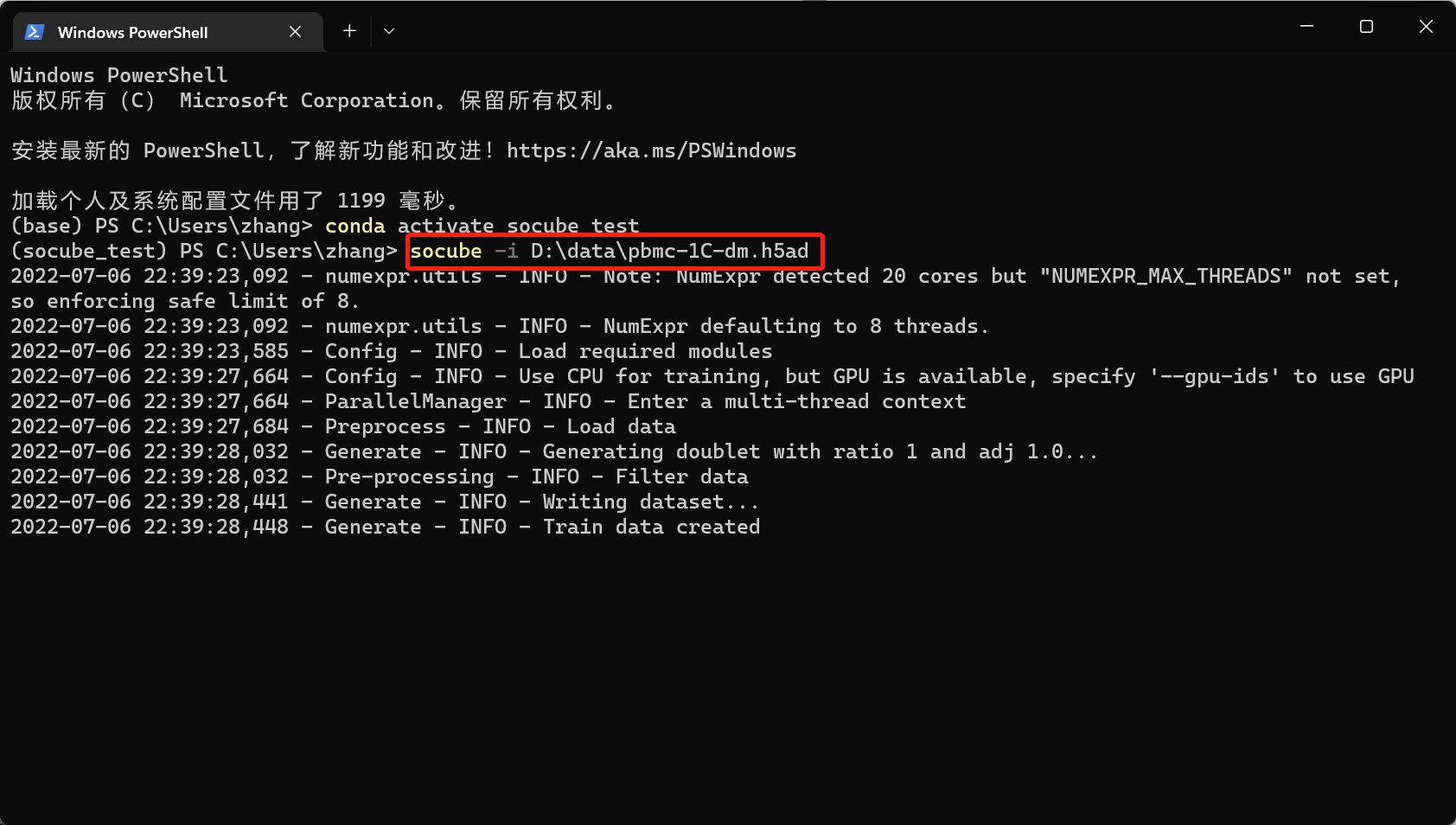
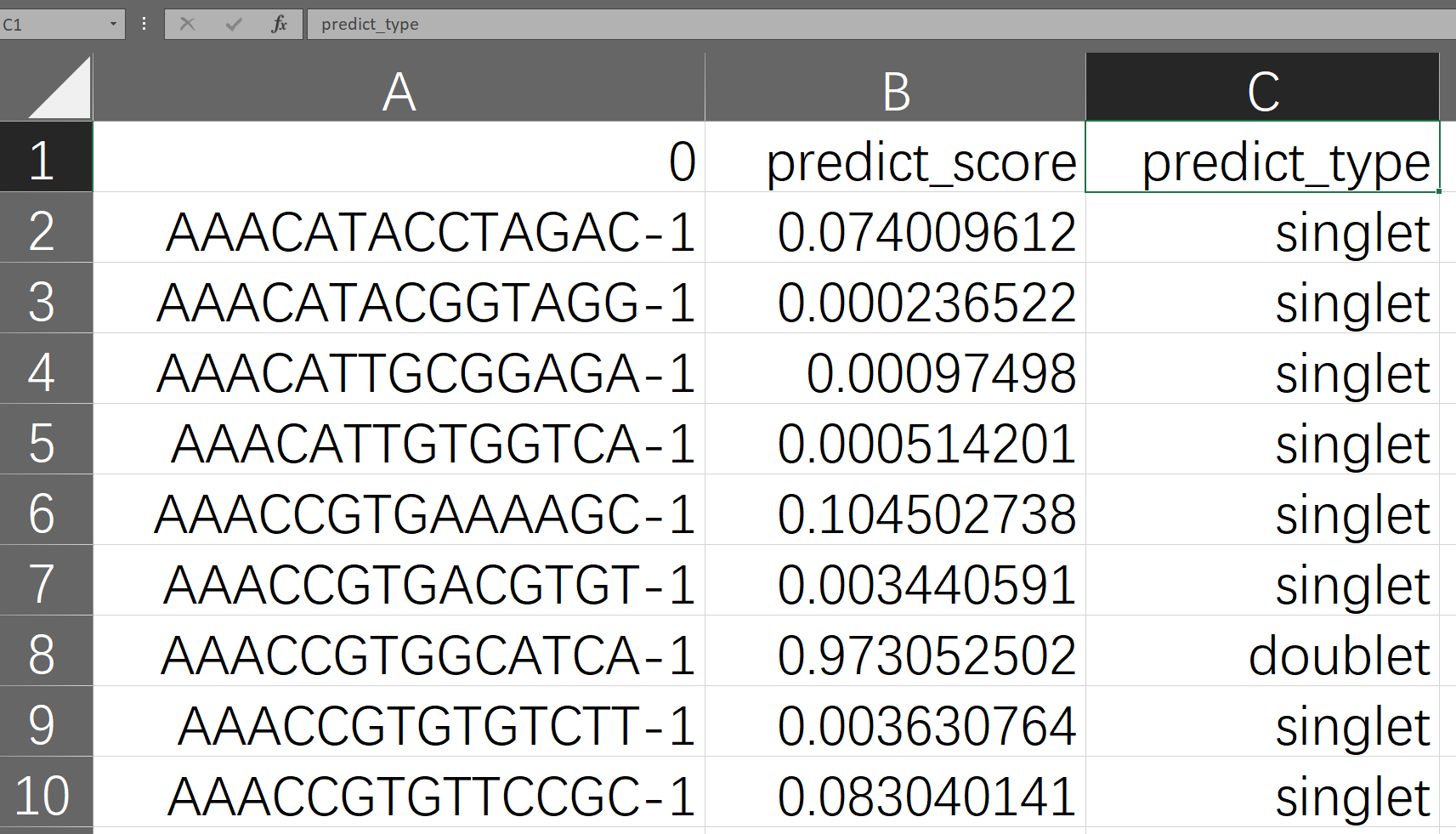
等待软件运行完毕,二聚体检测结果以及中间文件默认会和输入文件放在一起,产生embedding、plots、models、outputs这几个文件夹。其中最终结果在“outputs/你的模型ID/”文件夹下以“final_result”开头的CSV文件中。用Excel电子表格软件打开,可以看到其有下图三列。第一列是输入数据的细胞样本名称,由用户的输入决定;第二列是SoCube预测该样本为二聚体的概率值,大小介于0~1之间;第三列是SoCube基于用户给定概率阈值或者默认阈值0.5得出的类型预测,singlet即单体正常细胞,doublet即二聚体。用户既可以使用概率值进行后续自定义筛选,也可以利用第三列进行直接筛选。
4.2 GPU加速运算
如果用户拥有独立NVIDIA显卡并配置了正确的CUDA,会收到“Using CPU training, but GPU is available, specify ‘—gpu-ids’ to use GPU”的提示。用户只需要在前述命令的基础上,增加指定参数“—gpu-ids”来指定要使用的GPU。GPU的ID可以在前面说到的nvidia-smi命令输出中看到,0,1,2,…分别表示第1,2,3…块GPU(多块的GPU的情况下)
socube -i "D:\data\pbmc-1C-dm.h5ad" --gpu-ids "0,1"
4.3 基于docker的使用
和前面pip安装后使用本质上一样的,但需要通过docker来启动并进入容器,因此有额外步骤docker run启动容器以及docker exec进入容器。其中“-v”、“--gpus”,“--name”都是docker run命令的启动参数,“-v”参数将外部文件夹挂载到docker容器内部路径,请务必不要忘了加这个参数。因为socube读取的是docker容器内的文件路径。“--gpus”参数负责对容器使用gpu数量进行授权。进入容器后即可正常使用socube命令。
# 创建容器
docker run -v D:/data:/workspace/datasets `
–gpus all `
–name socube `
gcszhn/socube:latest
# 进入容器
docker exec -it socube bash# 创建容器
sudo docker run -v /data:/workspace/datasets \
–gpus all \
–name socube \
gcszhn/socube:latest
# 进入容器
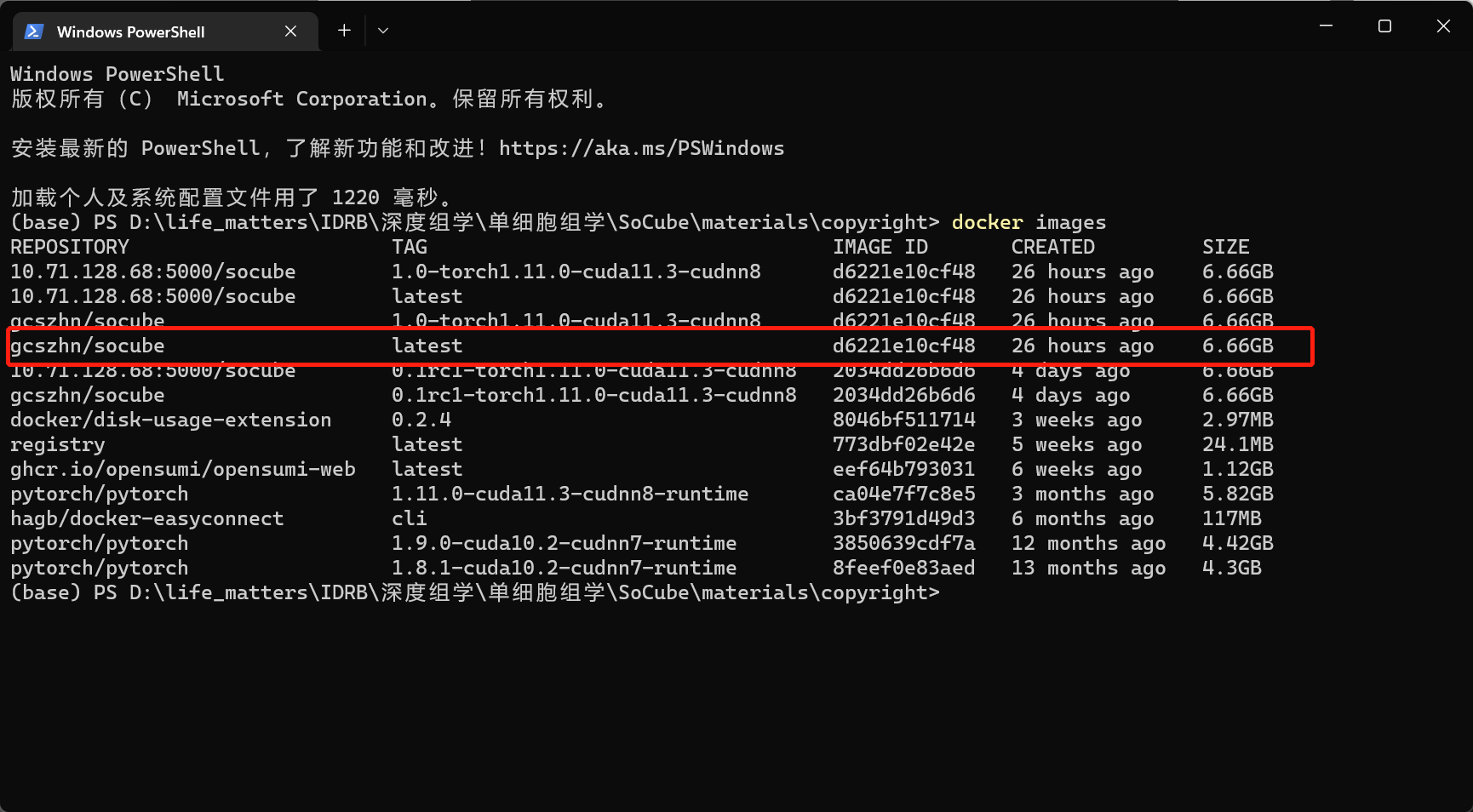
sudo docker exec -it socube bash其中“gcszhn/socube:latest”是镜像名称,前面的是docker启动参数,包括文件夹映射和GPU挂载,后面即SoCube的参数,使用与前文一致。用户可以通过docker images来查看镜像名称。
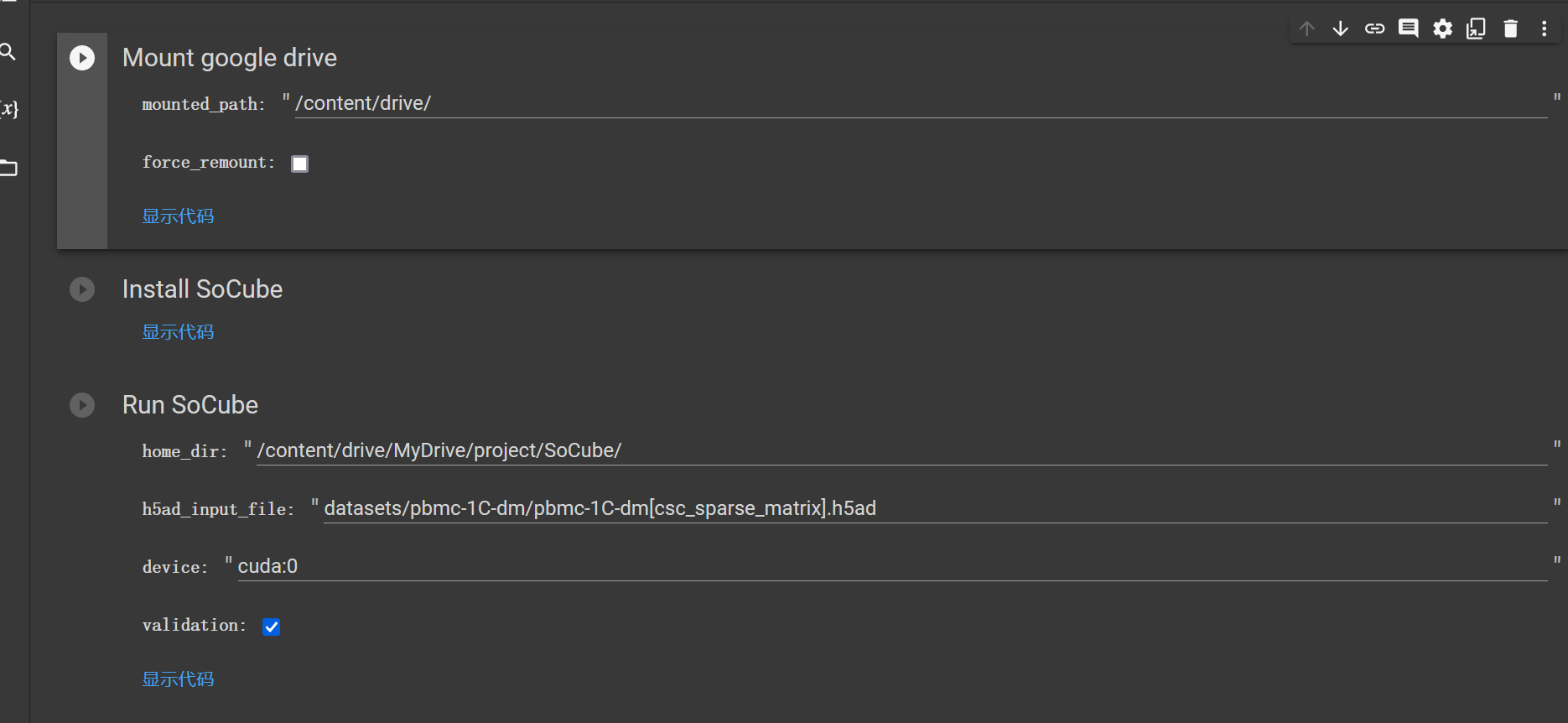
4.4 colab使用
谷歌提供了一个免费的带有GPU的在线机器学习平台colab。用户可以上传socube_colab.ipynb(在本项目的开源仓库中提供了)和scRNA-seq数据到你的google drive,并使用其进行GPU加速预测。(温馨提示:需要在notebook设置里面选择启用GPU,默认是CPU的)
4.5 多进程训练
在内存与显存都充足的情况下,用户可以使用--enable-multiprocess参数来启用多进程训练加速,充分发挥现代CPU的多核计算优势。
socube -i your_sc.h5ad -o your_sc --gpu-ids 0 --enable-multiprocess